Spinal Muscular Atrophy Causes
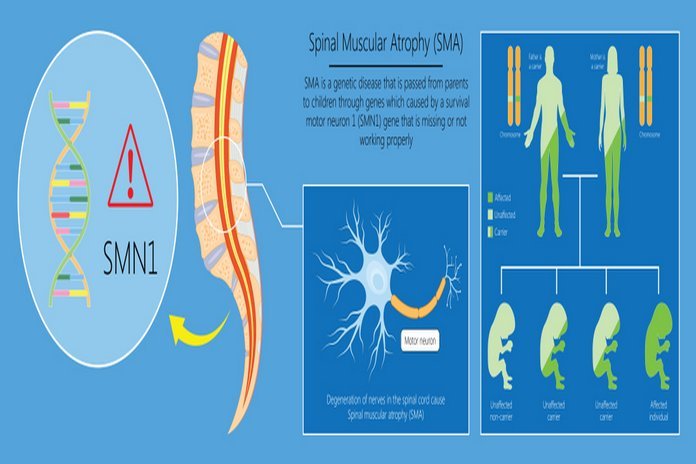
The most obvious and prevalent form of spinal muscular atrophy is the fault in both copies of the survival motor neuron one gene (SMN1) present on the 5q chromosome. SMN 1 gene is responsible for producing survival motor neuron SMN protein that maintains the motor neurons’ standard functions and health. Some individuals having insufficient levels of SMN protein develop spinal muscular atrophy. SMN protein, which is present in less amount, causes the loss of motor neurons in the spinal cord, leading to skeletal muscles’ wasting and weakness. The weakness resulting from spinal muscular atrophy is more severe in the limbs, i.e., upper leg and arm muscles and the trunk, then in the muscles of the feet and hand.
Several changes or mutations in the same genes become the cause of several types of spinal muscular atrophy. Some less common types of spinal muscular atrophy arise due to the changes in other genes such as the genes on chromosome 20, chromosome 14, and chromosome 9. The type of spinal muscular atrophy largely depends on the severity of muscle weakness and onset age. Still, there is always an overlapping situation between the types of SMN.
Genes and SMN
Spinal muscular atrophy usually occurs in an autosomal recessive way, which means that the individual who gets SMN carries two mutated genes. It is common to see that each patient suffering from SMN inherits each mutated gene from both parents. Although, in some rare cases, SMN occurs due to the mutations in the UBA1 gene. The people who carry only one mutated gene do not have any disorder symptoms but are carriers of the disease. As we know that SMN is an autosomal recessive disorder, so there is a possibility that it affects more than one person in the family.
Under normal conditions, the SMN1 genes produce fully functional and full-length SMN proteins necessary for the body’s neurological and muscular functions. When the SMN1 genes suffer from the mutations, there are insufficient levels of SMN proteins. Due to the mutation on chromosome 5, another gene in the neighboring location, i.e., the SMN2 gene, also suffers. The proteins produced from the SMN2 genes are in small percentages and are not functional at all. Moreover, the functionality of the SMN proteins remains only at 10-15%.
There are multiple copies of the SMN2 gene present in the human body. Typically there are zero to eight copies of SMN2 genes in each individual. The greater the number of copies of SMN2 genes in the body, the higher the number of functional SMN protein available in the body; thus, the disease symptoms are way milder. The severity of the disease also depends on the onset and severity of different influencing biological pathways. The biological modifiers such as ZPR1 protein and plastin three proteins indicate the disease’s onset or severity.